Tidyverse Practice
20 Apr 2023Clone the Assignment 4 Respository
You should be used to the routine now. Look for the Canvas announcment with the link. Click on the link to create the repo. Start a new project in R by cloning the repo. (See Tuesday’s lab for a refresher)
Welcome to the Tidyverse
Tidyverse is a collection of R packages that make data manipulation and processing easier.
I have designed a tutorial to introduce you to some of the key features of these packages. Please click on the following link to start the tutorial. Keep notes in a .Rmd file (or written notes) to keep track of what you learn. You will be required to turn this in.
You can access the first tutorial at http://malooflab.phytonetworks.org/apps/tidyverse-tutorial/
Alternate access in case the link above doesn’t work
If for some reason the link above does not work, then you can run the following code to install and run the tutorial on your computer or Jetstream2 instance.
First, open Firefox; the tutorial will eventually launch there.
Then, in R studio: NOT NEEDED UNLESS ABOVE LINK ISN’T WORKING
devtools::install_github("UCDBIS180L/BIS180LTutorials") # only needs to be done once
learnr::run_tutorial("Tidyverse_Introduction", package = "BIS180LTutorials") #do this every time you want to start the tutorial.
A tidyverse cheatsheet
Keep this cheatsheet handy!
IMPORTANT: Create a knittable .Rmd
To turn in this (and all subsequent) R assignments, you will need to knit your .Rmd and turn in the resulting .html (along with the .Rmd). When you knit a file R starts a new session. This means that you must include all code that is required. for example code that reads in files or creates objects.
Analyzing the BLAST results from Assignment 2
Now that you have some R fundamentals lets deploy them on the blast results from the previous BLAST lab.
Questions that we want to answer:
- Did the different ways of running blast result in different “hits”, even if the number of hits was similar?
- Did the different ways of running blast affect the length and percent identity of the matches?
- How can we subset our ~ 2500 unique hits to a smaller size suitable for evolutionary analysis?
Load tidyverse
library(tidyverse)
Download the files
Although you have your own files from Assignment 2, I’d like you to download my versions. This way we are all starting from the same files for this lab.
In R we can use the download.file method to download files from the web.
We will work with the word size 11 and word size 28 megablast files, the word size 11 blastn file, the tblastx file, and the word size 11 dc-megablast file.
It is OK to cut and paste the code below. In fact, please cut and paste the code below
download.file(url="https://bis180ldata.s3.amazonaws.com/downloads/Assignment3/blastout.WS28.tsv.gz",
destfile = "../input/blastout.mega.WS28.tsv.gz") # use this to put the file in a different directory
download.file(url="https://bis180ldata.s3.amazonaws.com/downloads/Assignment3/blastout.WS11.tsv.gz",
destfile = "../input/blastout.mega.WS11.tsv.gz") # use this to put the file in a different directory
download.file(url="https://bis180ldata.s3.amazonaws.com/downloads/Assignment3/blastout.task_blastn.tsv.gz",
destfile = "../input/blastout.task_blastn.WS11.tsv.gz") # use this to put the file in a different directory
download.file(url="https://bis180ldata.s3.amazonaws.com/downloads/Assignment3/blastout.tblastx.tsv.gz",
destfile = "../input/blastout.tblastx.tsv.gz") # use this to put the file in a different directory
download.file(url="https://bis180ldata.s3.amazonaws.com/downloads/Assignment3/blastout.task_dc-megablast.tsv.gz",
destfile = "../input/blastout.task_dc-megablast.WS11.tsv.gz") # use this to put the file in a different directory
If you are having trouble downloading the files, first make sure you are in the correct working directory
You will notice that I have compressed the files. The tblastx results file is 73MB in size uncompressed, but only 6.6MB compressed! One of the most common problems we run into as bioinformaticians is running out of disk space. Get in the habit of compressing files when you can. In this case, the compressed file is less than 10% of the original size! R will read compressed files just fine, no gunziping required.
Data import
The easiest way to bring data into R is from .csv (comma-separated values) or .tsv/.txt (tab-separated or space-separated values) files. Today we already have the file in almost the correct format. In the future if you need to import other data into R, both of these formats can be created in Excel (choose “Save As…” in Excel and select the correct format). The relevant R tidyverse import functions are read_tsv() and read_csv(). There also is a read_excel() function but use it with caution…
One bummer is that the columns names for the columns in these files are not in standard tsv format (in the first line, tab separated like the data), but instead are buried in a comment line. Let’s pull those out.
This is a bit tricky; the headers are on the fourth line of the file. I am telling R to read the first four lines of the file so that we can get the header first.
headers <- readLines("../input/blastout.mega.WS11.tsv.gz", n = 4)
headers <- headers[4] # only keep the 4th line
headers
## [1] "# Fields: query acc.ver, subject acc.ver, % identity, alignment length, mismatches, gap opens, q. start, q. end, s. start, s. end, evalue, bit score, subject title"
Now need to break this up so that each column name is a separate element of a vector.
headers <- headers %>%
str_remove("# Fields: ") %>% # get rid of the extraneous information
str_split(", ") %>% #split into pieces
unlist() %>% #convert to vector
make.names() %>% #make names the R can understand
str_replace(fixed(".."), ".") %>% # get rid of double .. in names.
str_replace("X.identity", "pct.identity") # "%" got replaced with X, we can do better.
headers
## [1] "query.acc.ver" "subject.acc.ver" "pct.identity" "alignment.length"
## [5] "mismatches" "gap.opens" "q.start" "q.end"
## [9] "s.start" "s.end" "evalue" "bit.score"
## [13] "subject.title"
OK that was a lot of new commands. If you want to see what each one of them does, run the first two lines (omitting the final %>%), then run the first three lines, etc. Note how the output changes with each command. You will learn more about the str_ commands in a later lab.
Now let’s read in the real data, and specify to use the headers object we just created as the column names. The comment="#" tells R to ignore any lines starting with #
megaWS11 <- read_tsv("../input/blastout.mega.WS11.tsv.gz", col_names=headers, comment="#")
megaWS28 <- read_tsv("../input/blastout.mega.WS28.tsv.gz", col_names=headers, comment="#")
blastnWS11 <- read_tsv("../input/blastout.task_blastn.WS11.tsv.gz", col_names=headers, comment="#")
dc_megaWS11 <- read_tsv("../input/blastout.task_dc-megablast.WS11.tsv.gz", col_names=headers, comment="#")
tblastx <- read_tsv("../input/blastout.tblastx.tsv.gz", col_names=headers, comment="#")
Look in the right hand pane (click on the “Environment” tab if needed) and you can see a brief description of the data. There are 12545 rows and 13 columns in the first file. It is important that you look at files after they have been read in with the head() and/or summary() functions to make sure that the data is as you expect.
head(blastnWS11) # first 6 rows
summary(blastnWS11)
You can see that summary() has provided some nice summary statistics for each numeric column.
Rather than working on each tibble separately it would be nice to have them together in object. You’ll see why shortly. bind_rows does just what it says. We first create a list of the data frames so that we can assign them names.
blast.results <- bind_rows(list(megaWS11=megaWS11,
megaWS28=megaWS28,
blastnWS11=blastnWS11,
dc_megaWS11=dc_megaWS11,
tblastx=tblastx),
.id="strategy")
head(blast.results)
Now we have it all in a single data frame and can summarize for each search strategy.
Exercise 1: What are the total number of hits for each search strategy? hint: start with the blast.results tibble and use group_by() and summarize_(). Do not type out the results manually, instead make sure that your code generates a table (that is included in the knitted output). You should need 1 to 3 lines of code for this and you should not need to run separate commands for the different search strategies.
Wow that is a lot of hits for tblastx. Let’s explore a bit more:
Exercise 2: Using the blast.results tibble, for each search strategy, calculate:
- Average alignment length
- Maximum alignment length
- Total alignment length (sum of all alignment lengths)
- Average percent identity
For full credit start withblast.resultsand only call thesummarizefunction once (you can have multiple arguments in thesummarizefunction). Your code should produce a table as output (you do not need to type the table manually).
Exercise 3:
We have multiple hits per subject and we want to focus on the unique hits. Make a new object called uniq.blast.results that retains the single longest alignment for each subject in each strategy. Hint: use one of the slice functions.
Did it work? If so we should get the same counts as in the Linux lab. Check it:
uniq.blast.results %>%
group_by(strategy) %>%
summarize(count=n())
If your counts don’t match you did something wrong.
Exercise 4: Repeat the summary from Exercise 2, but now on the unique hits. How do the results fit with your understanding of these different search strategies?
Exercise 5: For the full blast.results set (not the unique ones), answer the following questions for each search strategy. You do not need to type out the results so long as your knitted markdown has the answer output in a table.
- What are the total number of hits?
- What proportion of hits have an e-value of 0?
- What proportion of hits have a percent identity < 50?
- What proportion of hits have an E-value of 0 and have a percent identity less than 50?
hint: It is helpful to take advantage of the fact that the logical value TRUE also has the numeric value of 1, whereas false = 0. So, for example, if we wanted to count the numbers > 5 in the sequence of numbers from 1:10 you could use this code:
test <- 1:10 #create sequence of numbers
test # make sure it worked
test > 5 # illustrate logical result
sum(test>5) # number of entries whose value is > 5
hint You will want to use group_by() and summarize(). For full credit, your code should only call summarize once (but with multiple arguments).
Exercise 6: Why do you think tblastx is so different?
Set overlap
Are the ~ 2500 hits found by the different methods the same 2500? Let’s find out. We will display the data using an UpSet plot. This is kind of like a Venn diagram but better when you have more than three sets.
You will need to install the package. This only need to done once, so you do not need to include it in your .Rmd
# only needs to be run once per machine
install.packages("UpSetR")
But this should be in the .Rmd
library(UpSetR)
UpSetR wants a data frame with one row per sequence and one column for each strategy, and 1s or 0s indicating presence or absence of the sequence in each strategy.
R has a built-in function, table that does this for us. For example, consider Star Wars characters:
## # A tibble: 10 × 14
## name homeworld species gender height mass hair_color skin_color eye_color
## <chr> <chr> <chr> <chr> <int> <dbl> <chr> <chr> <chr>
## 1 Luke Skywalker Tatooine Human mascu… 172 77 blond fair blue
## 2 C-3PO Tatooine Droid mascu… 167 75 <NA> gold yellow
## 3 R2-D2 Naboo Droid mascu… 96 32 <NA> white, bl… red
## 4 Darth Vader Tatooine Human mascu… 202 136 none white yellow
## 5 Leia Organa Alderaan Human femin… 150 49 brown light brown
## 6 Owen Lars Tatooine Human mascu… 178 120 brown, gr… light blue
## 7 Beru Whitesun … Tatooine Human femin… 165 75 brown light blue
## 8 R5-D4 Tatooine Droid mascu… 97 32 <NA> white, red red
## 9 Biggs Darkligh… Tatooine Human mascu… 183 84 black light brown
## 10 Obi-Wan Kenobi Stewjon Human mascu… 182 77 auburn, w… fair blue-gray
## # … with 5 more variables: birth_year <dbl>, sex <chr>, films <list>, vehicles <list>,
## # starships <list>
If we want to count the number of characters of each species, we use table:
starwars %>%
select(species) %>%
table()
## .
## Aleena Besalisk Cerean Chagrian Clawdite
## 1 1 1 1 1
## Droid Dug Ewok Geonosian Gungan
## 6 1 1 1 3
## Human Hutt Iktotchi Kaleesh Kaminoan
## 35 1 1 1 2
## Kel Dor Mirialan Mon Calamari Muun Nautolan
## 1 2 1 1 1
## Neimodian Pau'an Quermian Rodian Skakoan
## 1 1 1 1 1
## Sullustan Tholothian Togruta Toong Toydarian
## 1 1 1 1 1
## Trandoshan Twi'lek Vulptereen Wookiee Xexto
## 1 2 1 2 1
## Yoda's species Zabrak
## 1 2
Similarly for gender:
starwars %>%
select(gender) %>%
table()
## .
## feminine masculine
## 17 66
Hollywood gender bias anyone?
If we want a two-way table then we pass two columns. I also convert to a data frame because we will need it in that form for plotting:
sw.table <- starwars %>%
select(species, gender) %>%
table() %>%
as.data.frame.matrix()
sw.table
## feminine masculine
## Aleena 0 1
## Besalisk 0 1
## Cerean 0 1
## Chagrian 0 1
## Clawdite 1 0
## Droid 1 5
## Dug 0 1
## Ewok 0 1
## Geonosian 0 1
## Gungan 0 3
## Human 9 26
## Hutt 0 1
## Iktotchi 0 1
## Kaleesh 0 1
## Kaminoan 1 1
## Kel Dor 0 1
## Mirialan 2 0
## Mon Calamari 0 1
## Muun 0 1
## Nautolan 0 1
## Neimodian 0 1
## Pau'an 0 1
## Quermian 0 1
## Rodian 0 1
## Skakoan 0 1
## Sullustan 0 1
## Tholothian 1 0
## Togruta 1 0
## Toong 0 1
## Toydarian 0 1
## Trandoshan 0 1
## Twi'lek 1 1
## Vulptereen 0 1
## Wookiee 0 2
## Xexto 0 1
## Yoda's species 0 1
## Zabrak 0 2
Exercise 7: Use the commands above to create the data frame below from uniq.blast.results and store it in an object called upset.table. Only the first 6 lines are shown.
## blastnWS11 dc_megaWS11 megaWS11 megaWS28 tblastx
## AC_000192 1 1 1 0 1
## AF075257 0 0 0 0 1
## AF220295 1 1 1 0 1
## AF353511 1 1 1 0 1
## AF391541 1 1 1 0 1
## AF391542 1 1 1 0 1
Now we can create an upset plot:
upset(upset.table)
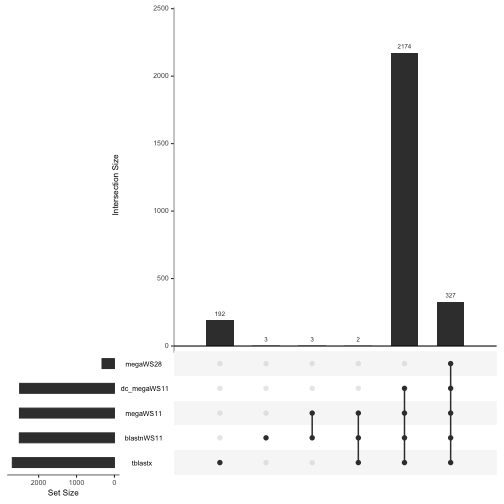
The rows represent the different strategies and the columns represent the different possible set memberships. Vertical bars show the number of sequences in each grouping. Dots and lines indicate the sets. So the left most column shows that there were 192 sequences unique to tblastx; the third column from the left indicates that there were 3 genes that were shared only between megaWS11 and blastnWS11
Exercise 8: Interpret the plot: overall do these strategies generally find the same sequences? Which strategy/ies are outliers? How does that relate to what you know about the different search strategies.
Filter the results
Finally, let’s filter this down to a reasonably sized set for alignment and phylogenetic tree reconstruction.
We will focus on blastnWS11. This has returned a good number of unique hits with long alignment lengths. Arguably we should include the tblastx results that are missed by blastnWS11 but to keep things somewhat simple let’s just use blastnWS11
First, create a data set that only has complete genome sequences from the blastnWS11 search
uniq.blastn <- uniq.blast.results %>%
ungroup() %>%
filter(strategy=="blastnWS11",
str_detect(subject.title, "complete genome"))
head(uniq.blastn)
## # A tibble: 6 × 14
## strategy query.acc.ver subject.acc.ver pct.identity alignment.length mismatches
## <chr> <chr> <chr> <dbl> <dbl> <dbl>
## 1 blastnWS11 Seq_H AC_000192 66.8 3938 1197
## 2 blastnWS11 Seq_H AF220295 65.8 5688 1739
## 3 blastnWS11 Seq_H AF353511 65.3 3200 982
## 4 blastnWS11 Seq_H AF391541 66.0 5682 1740
## 5 blastnWS11 Seq_H AF391542 65.9 5681 1744
## 6 blastnWS11 Seq_H AY274119 80.3 24666 4620
## # … with 8 more variables: gap.opens <dbl>, q.start <dbl>, q.end <dbl>, s.start <dbl>,
## # s.end <dbl>, evalue <dbl>, bit.score <dbl>, subject.title <chr>
IF you take a look at the subject.title from some of these, you realize that we have multiple isolates of the same virus:
uniq.blastn %>% pull(subject.title) %>% head()
## [1] "AC_000192 |Murine hepatitis virus strain JHM| complete genome|Murine coronavirus||"
## [2] "AF220295 |Bovine coronavirus strain Quebec| complete genome|Betacoronavirus 1||"
## [3] "AF353511 |Porcine epidemic diarrhea virus strain CV777| complete genome|Porcine epidemic diarrhea virus||"
## [4] "AF391541 |Bovine coronavirus isolate BCoV-ENT| complete genome|Betacoronavirus 1||"
## [5] "AF391542 |Bovine coronavirus isolate BCoV-LUN| complete genome|Betacoronavirus 1||"
## [6] "AY274119 |Severe acute respiratory syndrome-related coronavirus isolate Tor2| complete genome|Severe acute respiratory syndrome-related coronavirus|Canada|Homo sapiens"
We will want to filter based on some of these attributes, so first we need to split the subject.title column up into separate fields. We can do that with separate
uniq.blastn <- uniq.blastn %>%
separate(subject.title,
into=c("acc", "isolate", "complete", "name", "country", "host"),
remove = FALSE, #this keeps the "subject.title" column. We will need it later.
sep="\\|") # The | is a reserved search character and so we have to escape it with \\
## Warning: Expected 6 pieces. Additional pieces discarded in 1 rows [2081].
## Warning: Expected 6 pieces. Missing pieces filled with `NA` in 2 rows [1437, 2307].
# This will be explained in more detail in a later lesson.
head(uniq.blastn)
## # A tibble: 6 × 20
## strategy query.acc.ver subject.acc.ver pct.identity alignment.length mismatches
## <chr> <chr> <chr> <dbl> <dbl> <dbl>
## 1 blastnWS11 Seq_H AC_000192 66.8 3938 1197
## 2 blastnWS11 Seq_H AF220295 65.8 5688 1739
## 3 blastnWS11 Seq_H AF353511 65.3 3200 982
## 4 blastnWS11 Seq_H AF391541 66.0 5682 1740
## 5 blastnWS11 Seq_H AF391542 65.9 5681 1744
## 6 blastnWS11 Seq_H AY274119 80.3 24666 4620
## # … with 14 more variables: gap.opens <dbl>, q.start <dbl>, q.end <dbl>, s.start <dbl>,
## # s.end <dbl>, evalue <dbl>, bit.score <dbl>, subject.title <chr>, acc <chr>,
## # isolate <chr>, complete <chr>, name <chr>, country <chr>, host <chr>
Exercise 9: Let’s investigate those errors. Use the [ ] to view the offending rows of uniq.blastn. what went wrong?
uniq.blastn[c(1437, 2307, 2081),]
Exercise 10: Let’s delete those rows Use the [ ] to remove the offending rows of uniq.blastn. Put the result in a new object called uniq.blastn.filtered
End of Exercise 10
Next let’s eliminate rows with missing data. Missing data is currently indicated two different ways, in some cases the cell has an NA values and in other cases it is blank.
#first replace blank entries with NA
uniq.blastn.filtered <- uniq.blastn.filtered %>%
mutate_all(function(x) ifelse(x=="", NA, x))
# then remove any rows with an NA in them.
uniq.blastn.filtered <- na.omit(uniq.blastn.filtered)
nrow(uniq.blastn.filtered)
## [1] 2167
Check to see that you also have 2167 rows
Exercise 11: Look back to Exercise 3, where you created uniq.blast.results Use a similar strategy to retain the two entries with highest percent identity for each combination of name, country, and host in the uniq.blastn.filtered data frame. Put the results back into uniq.blastn.filtered.
Check it: you should have 492 rows
Exercise 12: Finally, filter to retain those with an alignment length >= 5000. Put the results back in to uniq.blastn.filtered
Check it: you should have 195 rows
Extract the sequences that we want.
OK! Let’s review our larger goals. We want to use the sequences in the uniq.blastn.filtered set to do a phylogenetic analysis. To do this we will need to create a new fasta sequence file that has the DNA sequences of interest, along with the relevant patient virus sequence. That is, we now need to subset the ncbi_virus_110119_2.txt fasta file to retain only the sequences listed in filtered.blastn
Luckily the Bioconductor package BioStrings makes it trivial to do this.
library(Biostrings)
We use the readDNAStringSet() function to read the fasta file into R. You will probably have to modify the path to point to your Assignment 2 repository and maybe remove the .gz from what I have below.
ncbi.seqs <- readDNAStringSet("../input/ncbi_virus_110119_2.txt.gz")
ncbi.seqs
## DNAStringSet object of length 122200:
## width seq names
## [1] 10705 GTCTACGTGGACCGACAAAGACAG...AAAATGGAATGGTGCTGTTGAAT MH781015 |Dengue ...
## [2] 198157 ATGGACAGCATAACTAACGGAGAA...TCGCAGTACACTACAAAGTTAAC NC_044938 |Heliot...
## [3] 193886 ACAATTTTATATTATAGTGCAGAT...TCTGCACTATAATATAAAATTGT NC_044944 |Africa...
## [4] 190773 TCTTATTACTACTGCTGTAGGCGT...CGCCTACAGCAGTAGTAATAAGA NC_044950 |Africa...
## [5] 34080 AATAAATAACGAAACATGCAAATT...ATTTGCATGTTTCGTTATTTATT NC_044960 |Bottle...
## ... ... ...
## [122196] 2585 CGGTGGCGTTTTTGTAATAAGAAG...TCTGAGAGAATCTATTTGTTAAA X15984 |Abutilon ...
## [122197] 2632 CGGTGGCATTTATGTAATAAGAAG...GCCTGTACTCTAAATTTCTTTGG X15983 |Abutilon ...
## [122198] 1291 CGCCAAAAACCTCTGCTAAGTCCC...ATCGGACGGCTGAGTTGATCTGG M29963 |Coconut f...
## [122199] 6355 GATGTTTTAATAGTTTTCGACAAC...CCCTAACCGCCGGTAGCGGCCCA M34077 |Tobacco m...
## [122200] 12297 GTATACGAGGTTAGCTCTTTCTCG...AAGGTAATTTCCTAACGGCCCCC J04358 |Classical...
BioStrings conveniently displays the first and last 5 sequences in the set (and only the first and last bases of each sequence) as a summary when we ask R to print the ncbi.seqs object.
Having loaded the sequences in to R, we want to select the subset of the sequences that we want to work with. We can do this with the [ ] square brackets and a vector of the sequence names that we want. This should seem familiar from swirl, but to review:
Example: I have a named vector of fruit sugar and I want to subset it to only those fruit that are currently in the store.
# fruit sugar content in g per cup of fruit
# Source: https://www.eatthis.com/fruit-sugar/
fruitSugar <- c(
"apples" = 13,
"blueberries" = 14.7,
"cherries" = 17.7,
"figs" = 29.3,
"kiwi" = 16.2,
"mango" = 22.5,
"plums" = 16.4,
"pomegranate seeds" = 23.8,
"raspberries" = 5.4,
"tangerines" = 20.6,
"watermelon" = 9.4
)
fruitsInStore <- c("apples", "blueberries", "mango", "tangerines")
Because this is a named list I can use the [ ] square bracket extractor to extract the names items that I want (fruit in the store)
fruitSugarSubset <- fruitSugar[fruitsInStore]
fruitSugarSubset
## apples blueberries mango tangerines
## 13.0 14.7 22.5 20.6
The same principle works on the sequence object.
Exercise 13: Use [ ] to subset the ncbi seqs to retain only those present in uniq.blastn.filtered
Hint: you will want to use the subject.title column from uniq.blastn.filtered.
Put the resulting sequences in an object named selected.seqs. You should have 195 sequences.
Exercise 14: Read in the patient seq file, extract the Seq_H sequence, and then add it to the selected.seqs object using c(). The new sequence object should have 196 sequences. Write it out to a fasta file using the function writeXStringSet(), naming the resulting file “selected_viral_seqs.fa”.
Exercise 15: Copy or move your tidyverse notes into the repo, add and commit them. Enter the file name(s) here: