Multiple sequence alignment and phylogenetic tree construction
27 Apr 2023Orientation
We would like to better examine the evolutionary origin of the virus causing the respiratory disease outbreak. In the last lab we used tools in R to narrow down our BLAST results to a manageable number of sequences and to create a new FASTA file that had just the sequences that we wanted.
Today we will do a multiple sequence alignment on those sequences and then build a phylogenetic tree.
Basic background on multiple sequence alignment and phylogenetic trees can be found in Chapter 9 of Bioinformatics for Beginners. You can access this through the library VPN.
Download the sequences
Just to make sure we are all starting with the same sequences, download my output from Assignment 4, part 1 and use these.
Download the sequences into your output folder with the code below. Make sure that your working directory is scripts, or, run this from within your assignment_4_template_2 script:
download.file(url="https://bis180ldata.s3.amazonaws.com/downloads/Assignment4/selected_viral_seqs_195v2.fa",
destfile = "../output/selected_viral_seqs_195v2.fa") # use this to put the file in a different directory
Multiple sequence alignment
There are many sequence alignment algorithms and programs. Here we will use MAFFT because it is reasonably quick and does a reasonably good job. Obtaining a good alignment is as much of an art as a science. I tried a few settings and found that we had to reduce the gap opening penalty to get a good alignment.
It takes about an hour to align (remember this is a big file, 196 sequences up to 24,000 bases in length). I give the command below, but YOU DO NOT NEED TO RUN THE COMMAND BELOW. IT WILL TAKE AN HOUR. DOWNLOAD MY RESULTS INSTEAD
#time mafft --maxiterate 100 --thread 3 --reorder --op 0.5 selected_viral_seqs_195v2.fa > mafft_maxiter100_195_op.5.fa
download.file(url="https://bis180ldata.s3.amazonaws.com/downloads/Assignment4/mafft_maxiter100_195v2_op.5.fa",
destfile = "../output/mafft_maxiter100_195v2_op.5.fa") # use this to put the file in a different directory
There is also a web interface at the MAFFT link above; if you use the web interface be sure to change the settings to match above. (I haven’t tested the web interface to see if it will run a file this big or not)
View alignment
It is always important to view your alignment to see if the aligner seems to have done a good job. There are many alignment visualization packages and websites. One that I like is Wasabi.
Go to Wasabi now to view the MAFFT output file. You can do this from your instance or from your own computer. You do not need to create a Wasabi account.
At Wasabi, click on the left most icon (document icon) to upload (import) a file. Select and upload the mafft_maxiter100_195v2_op.5.fa file
STOP and THINK: What is each row? What is each column? What are the colors?
Click on the “-“ icon to zoom out; I recommend zooming so far out that you just see colors, not letters. Scroll through the alignment. You will see that there are some regions that are aligned far better than others.
An example of a poorly aligned region:
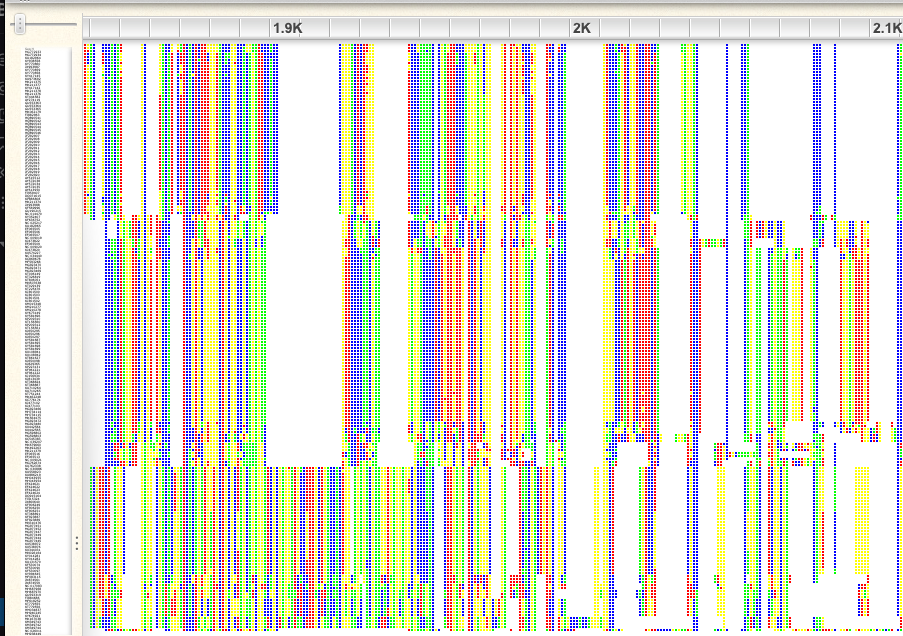
An example of a well-aligned region:

If you look at the first 1,000bp, you will see that there are some sequences that have gaps in otherwise very well conserved regions. A similar problem exists at the end of the alignment. Probably these are incomplete sequences, so we will plan to trim them. We will trim both of these regions, later in R.
There are also many gap-rich regions, where the alignment did not work well. We will also plan to trim these in R. You can get an idea for what removing gappy sites looks like by clicking on the gear icon in Wasabi and choosing “Hide gaps” and choosing a 75% threshold. This makes the missing data at the beginning and end of the alignment easier to see.
In theory we could do the trimming and export from Wasabi itself, but in practice that does not work well with this many sequences of this length, so we will turn to R.
Exercise 16:
If you turned on a gap threshold in the viewer, undo it by clicking on the undo button:
For each of the the following regions classify them as well- or poorly aligned, or uncertain/intermediate and explain your reasoning:
- 39.1K to 39.4K
- 31.6K to 31.9K
- 24.7K to 24.9K
- 14.5K to 14.7K
Some additional notes on alignments:
We are really doing a pretty quick and dirty job of editing of the alignment. If we were going to do this in more detail, we might use a program like gblocks to more systematically prune poorly aligned regions (although for the most part I think the 75% gap cutoff results in a reasonable alignment). An alternative approach would be to focus on a few genes that are conserved among these viruses, and then use an amino-acid or codon guided alignment using software like PRANK.
Subset sequences
Now that we have found a region we want to use for tree construction, let’s use R to subset those sequences.
RECOMMEND CUT and PASTE for this lab.
library(tidyverse)
library(Biostrings)
Load the alignment using Biostrings, and set up our file names.
inpath <- "../output/mafft_maxiter100_195v2_op.5.fa"
outpath <- "../output/mafft_maxiter100_195v2_op.5_trimmed_75pct.fa"
alignment <- readDNAMultipleAlignment(inpath)
alignment
Subset to trim off the ends with incomplete sequence
alignment <- DNAMultipleAlignment(alignment,start=1000,end=48449)
Mask sites that have more than 25% gaps
alignment <- maskGaps(alignment, min.fraction=0.25, min.block.width=1)
maskedratio(alignment) #what proportion got masked? (rows and columns)
## [1] 0.0000000 0.5212013
Change the alignment into a stringset so that we can more easily manipulate the names
alignment <- alignment %>% as("DNAStringSet")
For tree viewing we need to remove the “ “ (spaces) so that the names do not get truncated. We will also select a subset of fields. Many of these commands will be covered in a subsequent lab. OK to CUT and PASTE
newnames <- names(alignment) %>%
tibble(name=.) %>%
mutate(name=str_replace_all(name," ","_")) %>% #replace " " with "_" because some programs truncate name at " "
separate(name,
into=c("acc", "isolate", "complete", "name", "country", "host"),
sep="_?\\|_?") %>%
mutate(name=str_replace(name, "Middle_East_respiratory_syndrome-related","MERS"), # abbreviate
name=str_replace(name, "Severe_acute_respiratory_syndrome-related", "SARS"), # abbreviate
newname=paste(acc,name,country,host,sep="|")) %>% # select columns for newname
pull(newname) #return newname
## Warning: Expected 6 pieces. Missing pieces filled with `NA` in 1 rows [1].
head(newnames)
## [1] "Seq_H|NA|NA|NA"
## [2] "MG772933|SARS_coronavirus|China|Rhinolophus_sinicus"
## [3] "MG772934|SARS_coronavirus|China|Rhinolophus_sinicus"
## [4] "KU182964|Bat_coronavirus|China|Rhinolophus_ferrumequinum"
## [5] "KY938558|Bat_coronavirus|South_Korea|Rhinolophus_ferrumequinum"
## [6] "KY770860|Bat_coronavirus|China|Rhinolophus_ferrumequinum"
Write out the trimmed file
names(alignment) <- newnames
alignment %>% writeXStringSet(outpath)
Build a tree
Like everything else in this lab, there are a myriad of phylogenetic tree reconstruction algorithms and programs. There is a consensus that you want to use a Maximum Likelihood or Bayesian approach, not Maximum Parsimony and not Neighbor Joining.
We will use FastTree because it offers a good balance of accuracy and speed. Again this can be more of an art.
You can run FastTree from the command line or use the website.
At the LINUX COMMAND LINE and in your output directory, use:
FastTree -nt -gtr -gamma -out mafft_maxiter100_195v2_op.5_trimmed.fasttree.tre mafft_maxiter100_195v2_op.5_trimmed_75pct.fa
(Takes less than 2 minutes)
Where
-ntspecifies that this is a nucleotide alignment-gtrspecifies that we want to use the General Time Reversible model for nucleotide substitution-gammaimproves the maximum likelihood estimates-outspecifies what we want the output file to be named- the final argument is the input file.
Examine the tree
Again, there are many tree viewing programs. I like the web-based viewer iTOL.
Click on the Upload button at top of the screen.
Upload the mafft_maxiter100_195v2_op.5_trimmed.fasttree.tre file to iTOL.
It may take a few seconds for the file selection window to appear.
For improved viewing, do the following:
- Basic tab
- Mode: “Rectangular”
- Label options, Position: “At tips”
- Advanced tab
- Bootstraps / metadata: “Display”
- Legend: On
(Now the circles at each internal node indicate support for the node: 1 is best, 0 is worst).
You can now use the + and - buttons to make the tree larger and smaller. Clicking on the tree and scrolling allows you to move around.
Exercise 17:
There my be minor differences in the wording of the questions here vs the template. Use these
A: What is the sister taxon to Seq_H? (there will be two taxa in this group) What is the host for the virus in this group (provide the Latin and common names)
hint: if you are having trouble finding Seq H in the tree, search for it using the (Aa) magnifying glass
B: Consider Seq_H plus its sister taxa as defining one taxonomic group with three members. Look at the sister taxon of this group (it is a large group). What is a general description for the viruses in this sister group? List at least 3 different hosts found in this group. Give Latin and common names (if there is a common name).
C: Now consider Seq_H plus all of the other taxa used in question B as one taxonomic group. The sister to this group has three members. What are the hosts of this group?
D: Given your above findings, what do you think the host of the most recent common ancestor to the viruses in parts A and B was? (You can answer giving the common name for the general type of organism, e.g. rat, mouse, bat, ape, etc. you do not need to give a precise species).
E: Do you think that Seq_H evolved from a virus with a human host? Why or why not? If not, what did it evolve from?
Congratulations! You have identified the likely evolutionary origin of the COVID-19 virus.